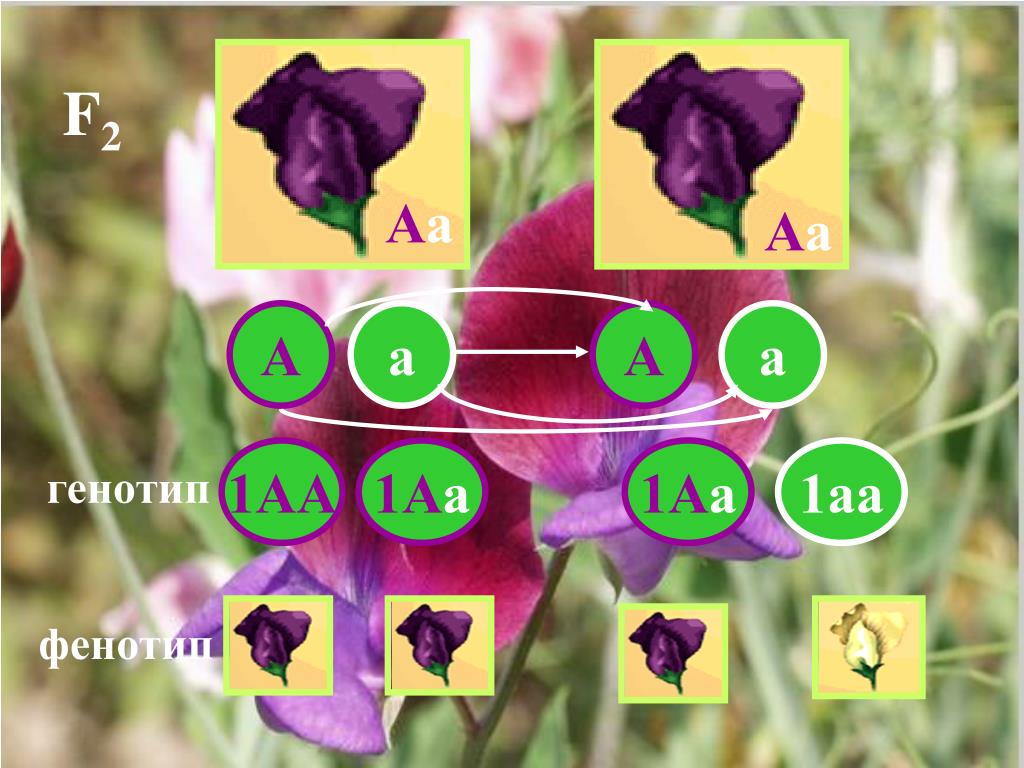
Понятие генотип и фенотип.
Генотип – совокупность наследственных признаков и свойств, полученных особью от родителей. А также новых свойств, появившихся в результате мутаций генов, которых не было у родителей. Генотип складывается при взаимодействии двух геномов (яйцеклетки и сперматозоида) и представляет собой наследственную программу развития, являясь целостной системой, а не простой суммой отдельных генов. Целостность генотипа – результат эволюционного развития, в ходе которого все гены находились в тесном взаимодействии друг с другом и способствовали сохранению вида, действуя в пользу стабилизирующего отбора. Так, генотип человека определяет (детерминирует) рождение ребенка, у зайца – беляка потомство будет представлено зайчатами, из семян подсолнечника вырастет только подсолнечник.
Генотип – это не просто сумма
генов. Возможность и форма проявления
гена зависят от условий среды.
Фенотип – совокупность всех признаков и свойств организма, сложившихся в процессе индивидуального развития генотипа. Сюда относятся не только внешние признаки (цвет кожи, волос, форма уха или нома, окраска цветков), но и внутренние: анатомические (строение тела и взаимное расположение органов), физиологические (форма и размеры клеток, строение тканей и органов), биохимические (структура белка, активность фермента, концентрация гормонов в крови). Каждая особь имеет свои особенности внешнего вида, внутреннего строения, характера обмена веществ, функционирования органов, т.е. свой фенотип, который сформировался в определенных условиях среды.
Понятия генотип и фенотип –
очень важные в генетике.
Известно, что генотип отражается в фенотипе, а фенотип наиболее полно проявляется в определенных условиях среды.
Норма реакции— способность генотипа формировать в онтогенезе, в зависимости от условий среды, разные фенотипы.
Термин введён в 1909 В. Иогансеном.
Она характеризует долю участия среды
в реализации признака. Чем шире норма
реакции, тем больше влияние среды и тем
меньше влияние генотипа в онтогенезе.
Один и тот же ген в разных условиях
среды может реализоваться в несколько
проявлений признака (фенов). В каждом
конкретном онтогенезе из спектра
проявлений признака реализуется только
один. Аналогично один и тот же генотип
в разных условиях среды может реализоваться
в целый спектр потенциально возможных
фенотипов, но в каждом конкретном
онтогенезе реализуется только один
фенотип.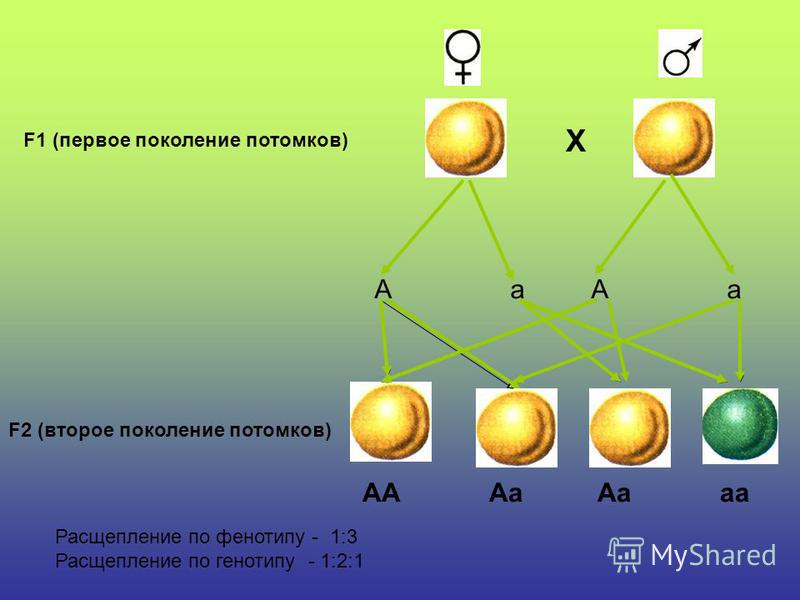
Под наследственной нормой реакции понимают максимально возможную ширину этого спектра: чем он шире, тем шире норма реакции.
Фенотипическое значение любого количественного признака определяется, с одной стороны, его генотипическим значением, с другой стороны — влиянием среды.
Широкая норма реакции: большие изменения признаков, например, надоев молока у коров, коз, массы животных.
Узкая норма реакции – небольшие изменения признаков, например, жирности молока, окраски шерсти.
Фактически норма реакции — спектр возможных уровней экспрессии генов, из которого выбирается уровень экспрессии, наиболее подходящий для данных условий окружающей среды.
Влияние генотипа и среды на фенотип
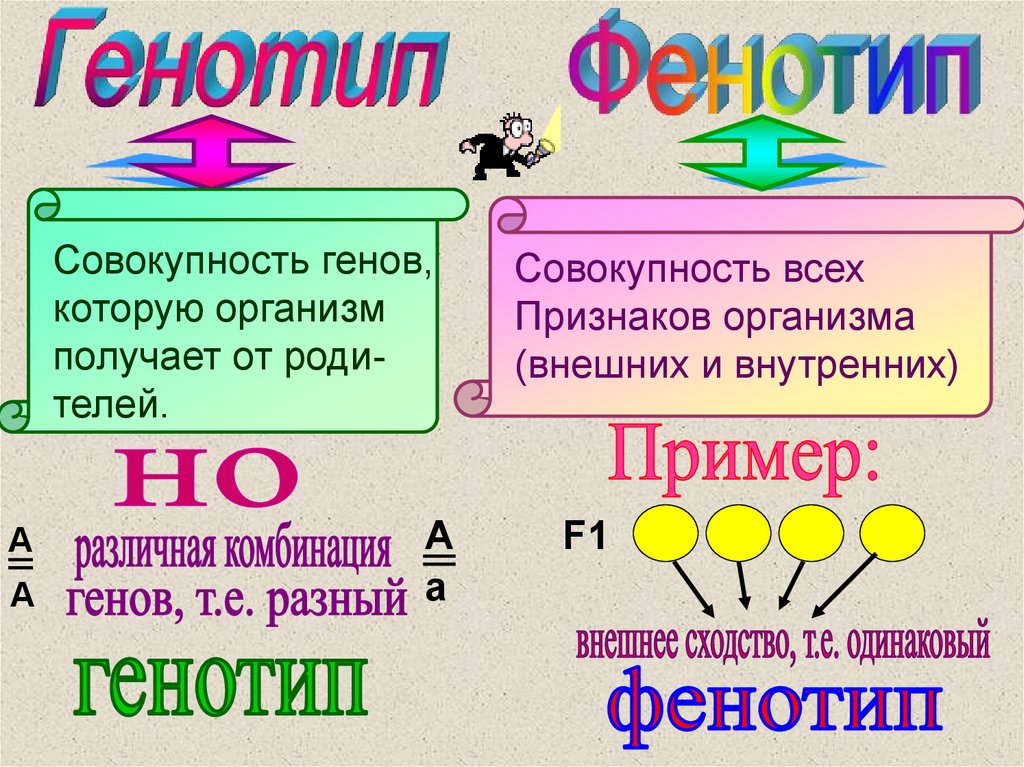
Напомним еще раз смысл понятий генотипа и фенотипа. Генотип — это
совокупность всех генов данного организма; фенотип — это совокупность всех
признаков организма.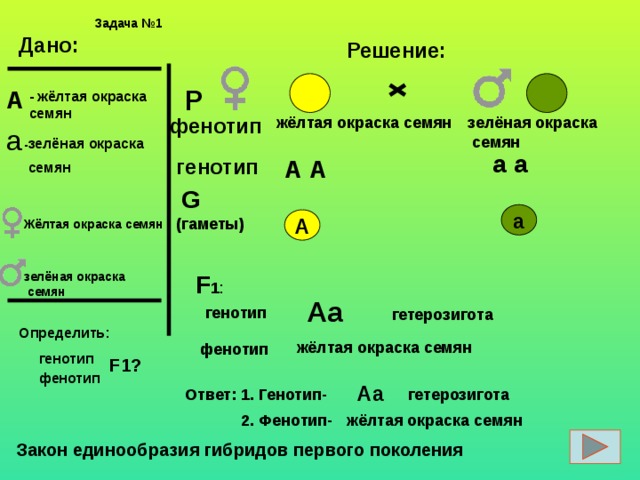
Известно, что при одном и том же фенотипе организмы могут иметь разный генотип. Например, в опытах Менделя растения, генотип которых содержал аллели AA, и растения, генотип которых содержал аллели Aа, по фенотипу не отличались друг от друга. Может ли быть обратная ситуация, когда генотипы у организмов одинаковые, а фенотипы разные? В частности, в какой мере фенотип определяется генотипом, а в какой — влияниями среды? Этот вопрос часто обсуждается на бытовом уровне применительно к характеру или поведению людей. При этом бытуют две точки зрения.
Согласно одной из них, особенности человека целиком определяются его генотипом. Поведение задано наследственностью, с которой ничего нельзя поделать. Согласно другой точке зрения, наследственность в поведении людей играет незначительную роль по сравнению с условиями жизни и, особенно, с воспитанием.
Рассмотрим влияние наследственности и среды на более простые признаки, чем
поведение людей. Даже для таких признаков возможны разные варианты.
Даже для таких признаков возможны разные варианты.
— Некоторые признаки полностью определяются генотипом и не зависят от условий среды. К ним относятся, например, группы крови и многие генетические заболевания .
— Другие признаки зависят и от генотипа и от среды. Например, рост человека зависит от его генотипа (вспомните работу Гальтона ). В то же время рост зависит и от условий среды, в частности от питания в период роста. Цвет кожи в значительной мере определяется генотипом. Но цвет кожи людей с одинаковым генотипом очень сильно зависит от времени их пребывания на солнце ( рис. 122 ).
Рассмотрим несколько характерных примеров влияния среды на проявления генов.
1. Еще на самом раннем периоде развития генетики было обнаружено, что
признак может оказаться
доминантным
или
рецессивным
в зависимости от условий, в которых развивается организм. В 1915 г. Морган показал на дрозофиле, что при выращивании в сухом воздухе обычное
для «дикого» типа распределение полос на брюшке дрозофилы
доминирует над ненормальным и, напротив, при избыточной влажности
доминирует ненормальное распределение полос. Наблюдения такого рода еще раз
показали различия между генотипом и фенотипом: при одном и том же генотипе
фенотип зависел от внешнцх условий.
Морган показал на дрозофиле, что при выращивании в сухом воздухе обычное
для «дикого» типа распределение полос на брюшке дрозофилы
доминирует над ненормальным и, напротив, при избыточной влажности
доминирует ненормальное распределение полос. Наблюдения такого рода еще раз
показали различия между генотипом и фенотипом: при одном и том же генотипе
фенотип зависел от внешнцх условий.
2. Влияние внешней среды на фенотип можно продемонстрировать на примере
общественных насекомых. У пчел и муравьев из неоплодотворенных яиц
развиваются самцы, а из оплодотворенных — самки. Однако фенотип этих самок
зависит от условий развития: при одних условиях развивается плодовитая
самка, а при других — бесплодная рабочая пчела. У
муравьев
существуют разные «касты» бесплодных особей. Основную часть
населения муравейника составляют рабочие муравьи, которые строят
муравейник, добывают пищу, выкармливают личинок и выполняют всякую другую
работу. У многих видов муравьев имеются «солдаты» — муравьи с
крупной головой, защищенной толстым хитином, и с особо мощными челюстями.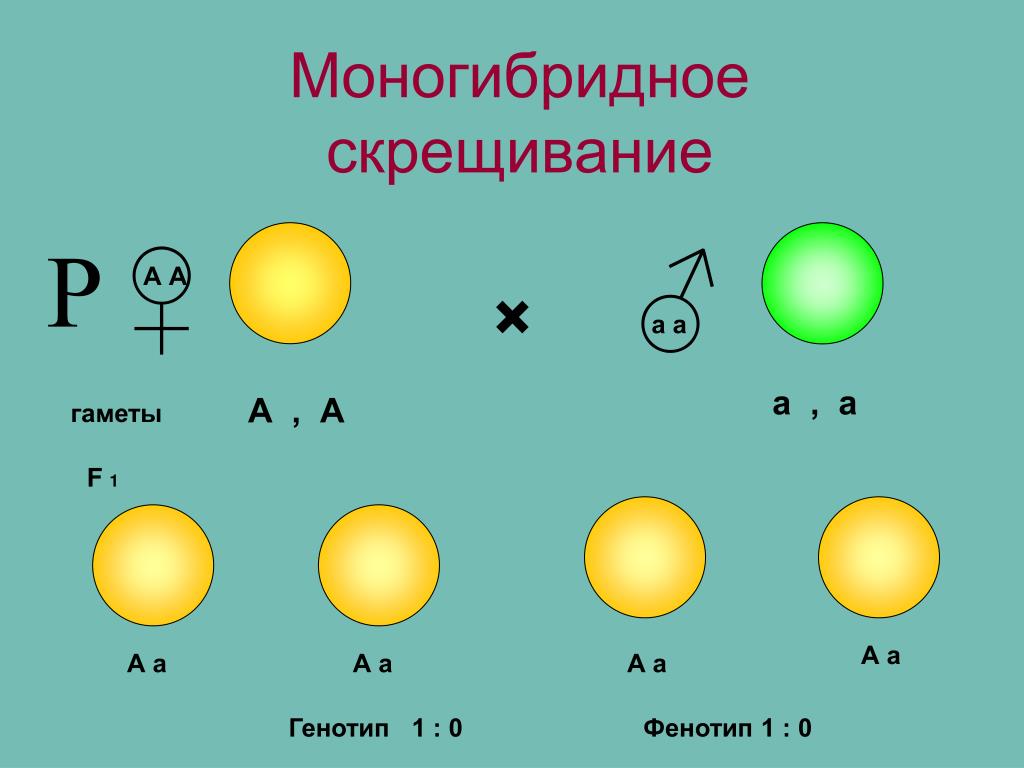
 Таким образом, было выяснено, что
развитие личинок зависит от того, какой корм они получат от рабочих
муравьев и какие добавки окажутся в корме. Точно так же у
пчел
от характера пищи и добавок зависит, разовьется личинка в рабочую пчелу
или же в матку.
Таким образом, было выяснено, что
развитие личинок зависит от того, какой корм они получат от рабочих
муравьев и какие добавки окажутся в корме. Точно так же у
пчел
от характера пищи и добавок зависит, разовьется личинка в рабочую пчелу
или же в матку.
3. У горностаевых кроликов мех белый, но отдельные части тела — лапы, уши, кончик морды и хвост — черные. Если на спине кролика, которая покрыта белой шерстью, выстричь какой-то участок и содержать кролика при пониженной температуре, на этом участке вырастает черная шерсть. Конечно, такие пятна черной окраски на необычном месте потомкам этого кролика по наследству не передаются.
Приведенные примеры показывают, что в действительности в очень многих случаях наследуется не признак как таковой, а способность к развитию данного признака при соответствующих условиях внешней среды, которая и передается от поколения к поколению.
Рассмотрим еще раз понятие
чистая линия
.
Датский генетик
Иоганнсен
поставил опыты для определения возможности отбора в чистых линиях. Он
видел, что у данного растения — гороха, принадлежащего к чистой линии,
имелись горошины разного размера: мелкие, средние и крупные. Иоганнсен
сажал самые мелкие горошины и самые крупные и получал от них потомство. С
растений, выросших из самых мелких горошин, брались опять самые мелкие, а с
растений, выросших из крупных горошин, брались самые крупные. После такой
процедуры, проводившейся для ряда поколений, оказалось, что соотношение
горошин разного размера (мелких, средних и крупных) было одинаковым у
отбираемых растений, выросших из самых мелких семян, и выросших из самых
крупных семян; при этом оно не отличалось от соотношения, которое было у
исходного родительского растения.
Вернемся теперь к поведению человека. Здесь возникают важные вопросы,
которые давно вызывают споры. Например, бывает ли человек от рождения умным
или глупым? Бывают ли врожденные преступники? Или ум — результат хорошего
воспитания, а преступность — результат плохого. Однако ответы на эти
вопросы очень трудны. Во-первых, трудно измерять уровень интеллекта
человека и характеристики его поведения. Во-вторых, трудно выяснить, какие
гены имеют отношение к поведению и как различаются люди по этим генам.
В-третьих, трудно сравнить или уравнять условия воспитания разных людей.
Во-вторых, трудно выяснить, какие
гены имеют отношение к поведению и как различаются люди по этим генам.
В-третьих, трудно сравнить или уравнять условия воспитания разных людей.
Тем не менее, некоторые результаты исследований этой проблемы заслуживают
внимания, например, полученные в работах по изучению наследования
интеллекта. Для определения уровня интеллекта разработан ряд тестов.
Применение этих тестов к близким родственникам, которые воспитывались
вместе или отдельно, и к неродственным людям, которые воспитывались вместе
или отдельно, показало следующее. Во-первых, чем ближе родство людей, тем
ближе их уровни интеллекта, даже если они воспитываются раздельно. Особенно
сходны между собой однояйцевые близнецы (Идея использования близнецов для
генетических исследований была предложена
Ф.Гальтоном
). Это значит, что генотип играет заметную роль в определении интеллекта.
Во-вторых, не родственники, воспитанные вместе, имеют более сходные
показатели интеллекта, чем такие же не родственники, воспитывающиеся
раздельно. Это показывает, что и среда (воспитание) отчасти определяет
уровень интеллекта. Для большинства людей влияние наследственности и среды
сопоставимо.
Это показывает, что и среда (воспитание) отчасти определяет
уровень интеллекта. Для большинства людей влияние наследственности и среды
сопоставимо.
Ссылки:
- Модификационная изменчивость
- ГЕНОТИП И ФЕНОТИП
От фенотипа к генотипу — PMC
- Список журналов
- Джей Дент Рез
- PMC4293721
Дж Дент Рез. 2014 июль; 93 (7 Дополнение): 3S–6S.
doi: 10.1177/0022034514533569
Геномика и трансформация первичной медико-санитарной помощи во всем мире
Информация об авторе Информация об авторских правах и лицензии Отказ от ответственности
За последние 50 лет был достигнут заметный прогресс в описании, измерениях и анализе фенотипов. Биомаркеры (белки, углеводы, липиды, гормоны, различные РНК и кДНК, микрочипы) были обнаружены и коррелированы с заболеваниями и расстройствами, а также с физиологическими реакциями на болезни, травмы, стрессы в крови, моче и слюне. Трехмерное цифровое изображение улучшило то, как мы «видим» и используем фенотипы для диагностики, лечения и прогноза. В каждом примере научное открытие привело к информированию клинического здравоохранения. В тандеме генетика развилась от менделевского наследования (мутации одного гена) до комплексных заболеваний человека (множественных взаимодействий ген-ген и ген-окружающая среда). Кроме того, эпигенетика расцвела новым пониманием генных модификаторов (например, метилирование гистонов и негистоновых хромосомных белков, ацетилирование, сульфатирование, фосфорилирование). Сейчас мы находимся в начале новой эры использования полногеномного секвенирования человека и микробов для принятия важных медицинских решений в отношении риска, стратификации пациентов, диагностики, лечения и результатов. Готовы ли мы как клиницисты, ученые и преподаватели к расширению сферы нашей практики, базы знаний, интеграции в первичную медико-санитарную помощь (медицина, фармацевтика, сестринское дело и смежные медицинские науки) и клинические подходы к черепно-лицевой, ротовой и стоматологической помощи? ? Время настало.
Трехмерное цифровое изображение улучшило то, как мы «видим» и используем фенотипы для диагностики, лечения и прогноза. В каждом примере научное открытие привело к информированию клинического здравоохранения. В тандеме генетика развилась от менделевского наследования (мутации одного гена) до комплексных заболеваний человека (множественных взаимодействий ген-ген и ген-окружающая среда). Кроме того, эпигенетика расцвела новым пониманием генных модификаторов (например, метилирование гистонов и негистоновых хромосомных белков, ацетилирование, сульфатирование, фосфорилирование). Сейчас мы находимся в начале новой эры использования полногеномного секвенирования человека и микробов для принятия важных медицинских решений в отношении риска, стратификации пациентов, диагностики, лечения и результатов. Готовы ли мы как клиницисты, ученые и преподаватели к расширению сферы нашей практики, базы знаний, интеграции в первичную медико-санитарную помощь (медицина, фармацевтика, сестринское дело и смежные медицинские науки) и клинические подходы к черепно-лицевой, ротовой и стоматологической помощи? ? Время настало.
Ключевые слова: персонализированная медицина и стоматология, полногеномное секвенирование, биоинформатика, эпигенетика, черепно-лицевые и рото-стоматологические заболевания и расстройства, феномика
Шестьдесят один год назад Уотсон и Крик опубликовали свою одностраничную статью о структуре ДНК (дезоксирибонуклеиновая кислота) с возможными биологическими последствиями (Watson and Crick, 1953). Препарат ДНК, который Розалинд Франклин использовала для своих исследований в области рентгеновской кристаллографии, был приготовлен молодым дантистом по имени Норман Симмонс. Тридцать девять лет назад я посетил семинар по рекомбинантной ДНК (19 лет).75) в Асиломаре, Калифорния, когда международное сообщество разработало, рекомендовало и приняло практические принципы. Двадцать шесть лет назад Джеймс Уотсон был назначен руководителем финансируемого из федерального бюджета проекта «Геном человека» (HGP). Год спустя его заменил Фрэнсис Коллинз, который довел HGP до завершения. В 2000 году президент Билл Клинтон просигнализировал о почти завершении HGP (Lander и др. ., 2001; Venter и др. ., 2001). В 2004 г. HGP был завершен в рамках бюджета и в срок; на это ушло 13 лет и 2,7 миллиарда долларов (Collins, 2010; Feero 9).0025 и др. ., 2010).
В 2000 году президент Билл Клинтон просигнализировал о почти завершении HGP (Lander и др. ., 2001; Venter и др. ., 2001). В 2004 г. HGP был завершен в рамках бюджета и в срок; на это ушло 13 лет и 2,7 миллиарда долларов (Collins, 2010; Feero 9).0025 и др. ., 2010).
В первые годы существования HGP (1988–2004 гг.) требовалось много месяцев или даже лет, чтобы завершить создание одного генома человека, что стоило миллионы долларов. Тем временем федеральные инвестиции вкладывались в биоинформатику, приборы и оборудование для высокопроизводительного секвенирования нуклеиновых кислот, чтобы ускорить и снизить стоимость секвенирования всего генома. В 2007 г. отдельные последовательности генома с аннотациями были завершены для Дж. Крейга Вентера и Джеймса Уотсона (Venter, 2007; Watson, 2009).). Это ознаменовало начало буквально персонализированного здравоохранения. В 2010 году Кевин Дэвис спроектировал индивидуальный геном стоимостью 1000 долларов и представил революцию в секвенировании ДНК и новую эру персонализированной медицины (и стоматологии) (Davies, 2010). Год назад Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) утвердило первую методологию и оборудование, способные завершить расшифровку генома человека в течение 24 часов при стоимости менее 5000 долларов США (Collins and Hamburg, 2013).
Год назад Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) утвердило первую методологию и оборудование, способные завершить расшифровку генома человека в течение 24 часов при стоимости менее 5000 долларов США (Collins and Hamburg, 2013).
Теперь мы знаем, что количество генетических вариаций между любыми двумя неродственными людьми составляет одно основание или нуклеотид (А, аденозин, Т, тимидин, С, цитозин и G, гуанозин) на тыс. оснований, или 0,1%. Каждый человек обладает вариациями примерно в 3 миллионах оснований из общего числа 3 миллиардов оснований, составляющих геном человека. Информация о каждой из последовательностей нашего генома, такая как однонуклеотидные полиморфизмы (SNP), теперь используется для понимания того, как вариации ДНК в нашем геноме могут влиять на наше здоровье (Kornman and Duff, 2012). Еще одно ключевое соображение для объяснения геномной изменчивости называется вариантами числа копий (CNV). Существует по меньшей мере 1500 CNV — сегментов хромосом, дублированных или утраченных у разных людей, — и они разбросаны по всему человеческому геному. Сейчас мы находимся в начале новой эры использования геномной информации для принятия важных медицинских решений для персонализированной стоматологии и медицины (Collins, 2010; Feero 9).0025 и др. ., 2010).
Сейчас мы находимся в начале новой эры использования геномной информации для принятия важных медицинских решений для персонализированной стоматологии и медицины (Collins, 2010; Feero 9).0025 и др. ., 2010).
Исторически сложилось так, что генетика изучает наследственность, процесс, при котором родитель передает определенные гены своим детям. Внешний вид или фенотип человека — рост, цвет волос, цвет кожи и цвет глаз — определяется генами (генотипами). Дополнительные характеристики или фенотипы, на которые влияет наследственность или спонтанные генетические мутации, включают метаболизм, умственные способности, природные таланты и восприимчивость или устойчивость к определенным заболеваниям или расстройствам.
После завершения HGP в 2004 году появилось гораздо более четкое понимание генома человека. Взрослые люди имеют десять триллионов соматических клеток, каждая из которых содержит 46 хромосом [2 половые хромосомы и 22 пары аутосомных хромосом (названы в соответствии с их размером, 1 самая большая и 22 самые маленькие)]. В геноме человека в ядре каждой соматической клетки тела содержится 21 000 функциональных генов и 19 000 псевдогенов. Незначительные вариации встречаются менее чем в 1% последовательности ДНК, что приводит к вариантам определенного гена, называемого аллелями (Feero 9).0025 и др. ., 2010). Гены представляют собой участки ДНК, а расположение гена называется локусом гена. Большинство функциональных генов кодируют информацию в экзонах, разделенных интронами, для создания белков. Каждый ген имеет расположенные выше последовательности энхансера и промотора, а также концевую стоп-последовательность. Каждый ген может производить более одного белка посредством процесса, называемого альтернативным сплайсингом. Кроме того, митохондрии внутриклеточных органелл также содержат ДНК (митДНК), унаследованную исключительно от наших матерей.
В геноме человека в ядре каждой соматической клетки тела содержится 21 000 функциональных генов и 19 000 псевдогенов. Незначительные вариации встречаются менее чем в 1% последовательности ДНК, что приводит к вариантам определенного гена, называемого аллелями (Feero 9).0025 и др. ., 2010). Гены представляют собой участки ДНК, а расположение гена называется локусом гена. Большинство функциональных генов кодируют информацию в экзонах, разделенных интронами, для создания белков. Каждый ген имеет расположенные выше последовательности энхансера и промотора, а также концевую стоп-последовательность. Каждый ген может производить более одного белка посредством процесса, называемого альтернативным сплайсингом. Кроме того, митохондрии внутриклеточных органелл также содержат ДНК (митДНК), унаследованную исключительно от наших матерей.
Несовершенный амелогенез, несовершенный дентиногенез, семейная агенезия зубов, синдром Папийона-Лефевра и серповидно-клеточная анемия — вот лишь несколько примеров из 10 000 заболеваний и нарушений, которые наследуются от мутаций одного гена через Менделевское наследование.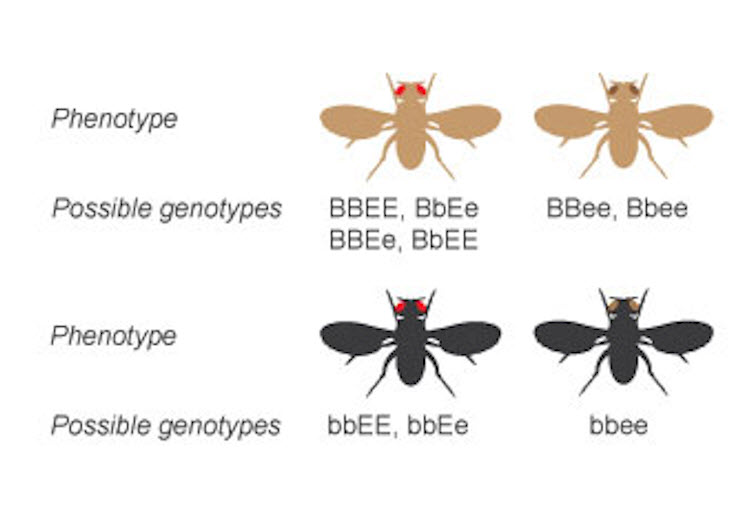 Известно более 60 наследственных заболеваний, возникающих в результате изменений (мутаций) митДНК, ассоциированных с рядом фенотипов, таких как слепота, тугоухость, низкорослость, метаболические нарушения. Подавляющее большинство заболеваний и расстройств человека являются полигенными и отражают множество взаимодействий ген-ген и ген-окружающая среда в сочетании с эпигенетическими модификаторами генов.
Известно более 60 наследственных заболеваний, возникающих в результате изменений (мутаций) митДНК, ассоциированных с рядом фенотипов, таких как слепота, тугоухость, низкорослость, метаболические нарушения. Подавляющее большинство заболеваний и расстройств человека являются полигенными и отражают множество взаимодействий ген-ген и ген-окружающая среда в сочетании с эпигенетическими модификаторами генов.
Любопытно, но мы знаем, что монозиготные близнецы имеют общий генотип. Имеют ли они общий фенотип? Недавно в нескольких исследованиях было установлено, что у монозиготных близнецов наблюдается фенотипическое несоответствие, такое как различия в восприимчивости к болезням, а также широкий спектр антропоморфных особенностей. В то время как монозиготные близнецы эпигенетически неразличимы в первые годы жизни, близнецы более старшего возраста демонстрируют значительные различия в распределении 5-метилцитозиновой ДНК и ацетилировании гистонов, что влияет на портрет экспрессии их генов. Короче говоря, многие новые исследования в настоящее время предоставляют доказательства того, что эпигенетика помогает понять различные фенотипы, которые могут происходить от одного и того же генотипа.
Короче говоря, многие новые исследования в настоящее время предоставляют доказательства того, что эпигенетика помогает понять различные фенотипы, которые могут происходить от одного и того же генотипа.
Еще одним аспектом геномики являются унаследованные, а также приобретенные мутации, например, обнаруживаемые при различных видах рака. Множественная эндокринная неоплазия 2 типа (МЭН 2) является примером аутосомно-доминантного наследственного ракового синдрома, вызванного миссенс-мутациями с усилением функции протоонкогена RET, и представляет собой сильные корреляции генотип-фенотип (Франк-Рауэ и Франк-Рауэ). , 2010). Повреждение окружающей среды канцерогенами или мутагенами, такими как содержащиеся в табачных изделиях и бензоле, являются примерами приобретенных мутаций в течение жизни, которые вызывают неоплазию, проявляющуюся при раке полости рта и глотки. Несколько различных генных мутаций, связанных с регуляцией клеточного цикла и внутриклеточных сигнальных сетей, вызывают различные виды рака. Полногеномное секвенирование образцов ДНК из биоптатов поражений пациента может быстро информировать об онкологических диагнозах и стратегиях химиотерапии (McDermott 9).0025 и др. ., 2011).
Полногеномное секвенирование образцов ДНК из биоптатов поражений пациента может быстро информировать об онкологических диагнозах и стратегиях химиотерапии (McDermott 9).0025 и др. ., 2011).
Три примера выбраны, чтобы подчеркнуть внедрение персонализированной медицины и стоматологии. Спустя более полувека после открытия молекулярной основы серповидно-клеточной анемии причины или объяснения фенотипической гетерогенности заболевания только проясняются. Серповидноклеточная анемия — это генетическое заболевание, при котором бета-цепь гена человеческого гемоглобина (Hb) мутирует, что приводит к аномальному гемоглобину. Эта мутация приводит к тому, что эритроциты (эритроциты) приобретают серповидную форму в условиях гипоксии, что приводит к целому ряду фенотипов, таких как анемия, клеточная адгезия, вазоокклюзия, сильная боль, инсульт и недостаточность органов. Генетическая мутация вызвана заменой одной аминокислоты глутаминовой кислоты на валин в шестом положении цепи бета-глобина. Это связано с заменой одного нуклеотида GAG → GTG в кодоне 6 гена бета-глобина, расположенного на хромосоме 11p15.5. Недавние исследования с использованием генотипирования SNP у пациентов с различными фенотипами выявили значительное участие SNP различных генов, отличных от бета-глобиновой цепи (Driss 9).0025 и др. ., 2009). SNP в генах, участвующих в путях трансформирующего фактора роста-бета/костного морфогенетического белка (TGF-бета/BMP), связаны с рядом фенотипических особенностей подмножеств у пациентов с серповидно-клеточной анемией (Driss et al. ., 2009).
Это связано с заменой одного нуклеотида GAG → GTG в кодоне 6 гена бета-глобина, расположенного на хромосоме 11p15.5. Недавние исследования с использованием генотипирования SNP у пациентов с различными фенотипами выявили значительное участие SNP различных генов, отличных от бета-глобиновой цепи (Driss 9).0025 и др. ., 2009). SNP в генах, участвующих в путях трансформирующего фактора роста-бета/костного морфогенетического белка (TGF-бета/BMP), связаны с рядом фенотипических особенностей подмножеств у пациентов с серповидно-клеточной анемией (Driss et al. ., 2009).
Диабет – это хроническое заболевание, определяемое гипергликемией. Различная степень резистентности к инсулину и/или дисфункция инсулин-продуцирующих бета-клеток поджелудочной железы вызывают диабет. Фенотип заболевания гетерогенен и может быть разделен на подтипы в зависимости от основной генетической причины (причин). Распространенные формы диабета — диабет 1-го типа (TIDM) и диабет 2-го типа (T2DM) — имеют значительную генетическую основу, но не имеют четкой картины наследования (Hornstein and Shuldiner, 2004).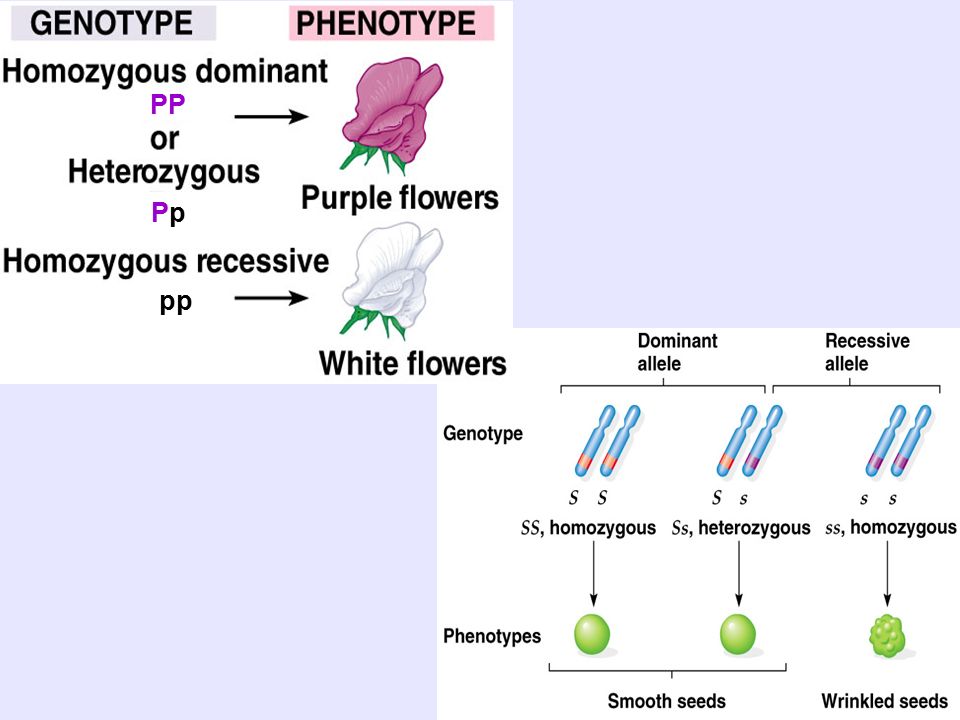 Фенотипы болезни возникают в результате взаимодействия нескольких вариантов генов с факторами окружающей среды. Следовательно, проведение генетических исследований в нескольких популяциях людей может выявить аллели риска заболевания, которые распространены в одной популяции, но чрезвычайно редки в других. Этот подход может пролить свет на патофизиологию, различия в состоянии здоровья и популяционно-генетическое происхождение аллелей болезней. Недавнее исследование показало, что варианты последовательности аллеля SLC16A11 являются частыми факторами риска развития диабета 2 типа в Мексике, но чрезвычайно редки в популяциях Европы и Африки (Williams 9).0025 и др. ., 2013). Продукты гена SLC16A11 изменяют метаболизм липидов и вызывают внутриклеточное увеличение метаболизма триацилглицеринов (Williams et al. ., 2013). Любопытно, что пациенты с плохо контролируемым диабетом (, т.е. , люди с риском развития ретинопатии, невропатии и макрососудистых заболеваний) могут быть предрасположены к пародонтиту с прогрессирующей потерей соединительной ткани и альвеолярной кости (Pihlstrom et al , 2005).
Фенотипы болезни возникают в результате взаимодействия нескольких вариантов генов с факторами окружающей среды. Следовательно, проведение генетических исследований в нескольких популяциях людей может выявить аллели риска заболевания, которые распространены в одной популяции, но чрезвычайно редки в других. Этот подход может пролить свет на патофизиологию, различия в состоянии здоровья и популяционно-генетическое происхождение аллелей болезней. Недавнее исследование показало, что варианты последовательности аллеля SLC16A11 являются частыми факторами риска развития диабета 2 типа в Мексике, но чрезвычайно редки в популяциях Европы и Африки (Williams 9).0025 и др. ., 2013). Продукты гена SLC16A11 изменяют метаболизм липидов и вызывают внутриклеточное увеличение метаболизма триацилглицеринов (Williams et al. ., 2013). Любопытно, что пациенты с плохо контролируемым диабетом (, т.е. , люди с риском развития ретинопатии, невропатии и макрососудистых заболеваний) могут быть предрасположены к пародонтиту с прогрессирующей потерей соединительной ткани и альвеолярной кости (Pihlstrom et al , 2005). ).
).
Заболевания пародонта широко распространены и поражают более 70% населения мира. Это заболевание приводит к значительной потере соединительной ткани и потере костной массы, связанной с потерей зубов у взрослых. В дополнение к различным микроорганизмам, образующим биопленку, этому заболеванию способствуют генетические факторы и факторы окружающей среды, особенно употребление табака. Генетические, дерматологические, гематологические, гранулематозные, иммуносупрессивные и опухолевые нарушения связаны с пародонтальными проявлениями (Pihlstrom 9).0025 и др. ., 2005). Дополнительные сведения получены в результате открытия того, что четыре различные мутации в экзонах гена катепсина С, обнаруженные на хромосоме 11q14, ответственны за синдром Папийона-Лефевра (Hart et al. ., 1999). Другие генетические нарушения с проявлениями заболеваний пародонта включают семейную и циклическую нейтропению, болезнь клеток Лангерганса, синдромы Чедиака-Хигаси, Элерса-Данлоса, Марфана, Дауна и Киндлера.
Полногеномный скрининг может позволить специалистам в области черепно-лицевого, орального и стоматологического здоровья выявлять и стратифицировать пациентов с высоким риском заболеваний пародонта (Giannobile et al. ., 2013a,b) и многие другие черепно-лицевые-орально-стоматологические заболевания и расстройства ( например ., черепно-лицевые синдромы, травмы головы и шеи, кариес зубов, рак головы и шеи, аутоиммунные заболевания, такие как как синдром Шегрена, остеопороз, височно-нижнечелюстные дисфункции, хроническая лицевая боль, остеопороз и др.). Существует значительная возможность использовать международное сотрудничество по теме взаимодействия генотип-фенотип. Такие усилия могут способствовать открытию специфических генов и сложных генных сетей, которые идентифицируют генотипы субпопуляций, подверженных риску, способствуют точной диагностике и разработке терапевтических средств, нацеленных на конкретные генотипы пациентов (Slavkin, 2014; Slavkin and Santa Fe Group, 2014).
Ближайшее будущее сферы ухода за полостью рта в Соединенных Штатах и странах по всему миру является неопределенным (DePaola and Slavkin, 2004; Glick, 2009; Frenk et al. ., 2010). Мы все больше осознаем, что реформы здравоохранения с различными вариантами возникали и возникают в разных штатах США и за их пределами (Glick, 2009; Frenk et al ., 2010). Несколько исследований рекомендовали пересмотреть традиционные кадры первичной медико-санитарной помощи, чтобы лучше справляться с болезнями и расстройствами в обществе (Frenk и др. ., 2010). Несколько известных групп выступили за то, чтобы геномика, фармакогеномика и иммуногеномика, а также микробиом, связанный с состоянием человека, стали частью образования и компетенций специалистов по гигиене полости рта (Collins and Tabak, 2004; Johnson et al. ., 2008; Глик, 2009; Славкин, 2012в).
С конца 1930-х годов многие специалисты в области гигиены полости рта начали заниматься генетикой человека и рекомендовали включить ее в качестве обязательной компетенции в стоматологическое образование, а также создать и поддерживать мультидисциплинарные команды, такие как черепно-лицевые группы, для удовлетворения особых потребностей головных врачей. врожденные дефекты шеи и шеи, травмы и рак (Купер, 19 лет).42, 1953; Славкин, 2012а,б; Фокс и Стоун, 2013 г.; Славкин и др. , в печати). Говоря современной терминологией, такие межпрофессиональные группы по образованию и здравоохранению (IPE) могут значительно улучшить глубину, широту и качество комплексной и скоординированной медицинской помощи на протяжении всей жизни.
врожденные дефекты шеи и шеи, травмы и рак (Купер, 19 лет).42, 1953; Славкин, 2012а,б; Фокс и Стоун, 2013 г.; Славкин и др. , в печати). Говоря современной терминологией, такие межпрофессиональные группы по образованию и здравоохранению (IPE) могут значительно улучшить глубину, широту и качество комплексной и скоординированной медицинской помощи на протяжении всей жизни.
Клиницисты и ученые-члены Международной ассоциации стоматологических исследований (IADR), Американской ассоциации стоматологических исследований (AADR), Национальных институтов здравоохранения (NIH) и многих других биомедицинских исследовательских организаций по всему миру участвуют в открытии фундаментальные знания о природе и поведении живых людей и систем и применять эти знания для улучшения условий жизни человека. Наука и научные открытия меняют жизнь каждого из нас. Научные открытия информируют клиническое здравоохранение! Мы должны инвестировать в геномику для улучшения клинической гигиены полости рта в 21 веке для всех людей (Славкин, 2012a,b,c)!
Автор хотел бы отметить новаторские и доблестные усилия доктора. Герберту Куперу, Саю Крешоверу, Сэму Прузански, Карлу Уиткопу, Бобу Горлину, Рэю Стюарту, Майклу Коэну-младшему, Тому Харту, Бобу Дженко, Чаку Шулеру и Ларри Табаку за продвижение генетики в рамках образования и клинической практики специалистов по гигиене полости рта.
Герберту Куперу, Саю Крешоверу, Сэму Прузански, Карлу Уиткопу, Бобу Горлину, Рэю Стюарту, Майклу Коэну-младшему, Тому Харту, Бобу Дженко, Чаку Шулеру и Ларри Табаку за продвижение генетики в рамках образования и клинической практики специалистов по гигиене полости рта.
Автор не получал финансовой поддержки и заявляет об отсутствии потенциального конфликта интересов в отношении авторства и/или публикации этой статьи.
- Коллинз ФС. (2010). Язык жизни: ДНК и революция в персонализированной медицине. Нью-Йорк, штат Нью-Йорк: Издательство Harper. [Академия Google]
- Коллинз Ф.С., Гамбург, Массачусетс. (2013). Первая авторизация FDA для секвенатора следующего поколения. N Engl J Med 369:2369-2371. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Коллинз Ф.С., Табак Л.А. (2004). Призыв к расширению образования в области генетики для специалистов в области стоматологии. Джей Дент Эдук 68:807-808. [PubMed] [Академия Google]
- Купер ХК. (1942).
 Дети-калеки?
Am J Orthod Oral Surg
28:35-40. [Академия Google]
Дети-калеки?
Am J Orthod Oral Surg
28:35-40. [Академия Google] - Купер ХК. (1953). Интеграция услуг в лечении расщелины губы и неба. J Am Dent Assoc. 47:27-35. [PubMed] [Академия Google]
- Дэвис К. (2010). Геном за 1000 долларов: революция в секвенировании ДНК и новая эра персонализированной медицины. Нью-Йорк, штат Нью-Йорк: Свободная пресса. [Академия Google]
- ДеПаола Д, Славкин ХК. (2004). Реформирование образования в области стоматологического здоровья: информационный документ. Джей Дент Эдук 48:1139-1350. [PubMed] [Академия Google]
- Дрисс А., Асаре К.О., Хибберт Дж.М., Джи Б.Е., Адамкевич Т.В., Стайлз Дж.К. (2009). Серповидноклеточная анемия в постгеномную эру: моногенное заболевание с полигенным фенотипом. Геномные идеи 2:23-48. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Feero WG, Guttmacher AE, Collins FS. (2010)Геномная медицина — обновленный учебник для начинающих. New Engl J Med. 362:2001-2011. [PubMed] [Академия Google]
- Фокс Л. М., Стоун П.А. (2013). Изучение командного процесса: разработка и поддержание эффективной черепно-лицевой помощи в команде. В: Расщелина губы и неба: диагностика и лечение. Берковиц С., редактор. Берлин, Германия: Springer-Verlag. [Академия Google]
- Франк-Рауэ К., Франк-Рауэ Р.С. (2010). Молекулярная генетика и феномика мутаций RET: влияние на прогноз МРЩЖ. Мол Селл Эндокринол 322:2-7. [PubMed] [Академия Google]
- Френк Дж., Чен Л., Бхутта З.А., Коэн Дж., Крисп Н., Эванс Т. и др. (2010). Медицинские работники для нового века: преобразование образования для укрепления систем здравоохранения во взаимозависимом мире. Ланцет 376:1923-1958. [PubMed] [Академия Google]
- Giannobile WV, Braun TM, Caplis AK, Doucette-Stamm L, Duff GW, Kornman KS. (2013а). Стратификация пациентов для профилактической помощи в стоматологии. Джей Дент Рез 92:694-701. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Джаннобиле В.В., Корнман К.С., Уильямс Р.С. (2013б). Персонализированная медицина входит в стоматологию: что это может означать для клинической практики?
J Am Dent Assoc
144:874-876. [PubMed] [Академия Google]
- Глик М. (2009). Расширение роли стоматолога в оказании медицинской помощи: не пора ли отказаться от прокрустово ложа? J Am Dent Assoc 140:1340-1342. [PubMed] [Академия Google]
- Hart TC, Hart PS, Bowden DW, Michalec MD, Callison SA, Walker SJ, et al. (1999). Мутации гена катепсина С ответственны за синдром Папийона-Лефевра. Джей Мед Жене 36:881-887. [Бесплатная статья PMC] [PubMed] [Google Scholar]
- Хорнштейн РБ, Шульдинер АР. (2004). Генетика диабета. Rev Endocr Metab Disord 5:25-36. [PubMed] [Академия Google]
- Джонсон Л., Дженко Р.Дж., Дамский С., Хейден Н.К., Харт С., Харт Т.С. и др. (2008). Генетика и ее значение для клинической стоматологической практики и образования: отчет группы 3 исследования Macy. Джей Дент Эдук 72 (2 Доп): 86-94. [PubMed] [Google Scholar]
- Корнман К.С., Дафф Г.В. (2012). Персонализированная медицина: оседлает ли стоматология волну или понаблюдает с пляжа?
Джей Дент Рез
91(7 Дополнение):8S-11S. [PubMed] [Академия Google]
- Ландер Э.С., Линтон Л.М., Биррен Б., Нусбаум С., Зоди М.С., Болдуин Дж. Международный консорциум по секвенированию генома человека и др. (2001). Первоначальное секвенирование генома человека. Природа 409:860-921. [PubMed] [Академия Google]
- Макдермотт У., Даунинг Дж. Р., Стрэттон М. Р. (2011). Геномика и континуум лечения рака. N Engl J Med 364:340-350. [PubMed] [Академия Google]
- Пихлстром Б.Л., Михалович Б.С., Джонсон Н.В. (2005). Заболевания пародонта. Ланцет 366:1809-1820. [PubMed] [Академия Google]
- Славкин ХК. (2012а). Рождение дисциплины: черепно-лицевая биология. Ньютаун, Пенсильвания: Aegis Communications. [Академия Google]
- Славкин ХК. (2012б). Ответ кроется в геноме. Глобальная связь здравоохранения 14:6-15. [Академия Google]
- Славкин ХК. (2012с). Эволюция научной основы стоматологии: с 1936 г. по настоящее время и ее влияние на стоматологическое образование. Джей Дент Эдук 76:28-35. [PubMed] [Академия Google]
- Славкин ХК. (2014). Будущее исследований в области черепно-лицевой биологии и что это будет означать для профессионального образования и клинической практики в области гигиены полости рта. Ост Дент Дж.
59(1 доп): 1С-5С. [PubMed] [Академия Google]
- Славкин ХК, Группа Санта-Фе (2014). Пересмотр сферы деятельности специалистов по гигиене полости рта. J Am Dent Assoc 145:228-230. [PubMed] [Академия Google]
- Славкин Х.К., Санчес-Лара П., Чай Ю., Урата М. (2014). Модель межпрофессиональной медицинской помощи: уроки, извлеченные из черепно-лицевых команд. J Calif Dent Assoc (в печати). [PubMed] [Академия Google]
- Вентер Дж. (2007). Расшифрованная жизнь: мой геном. Нью-Йорк, штат Нью-Йорк: Penguin Books. [Академия Google]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. (2001). Последовательность генома человека. Наука 291:1304-1351. [PubMed] [Академия Google]
- Уотсон Дж. (2009). Живу со своим личным геномом. Персонализированная медицина
6:607-610. [PubMed] [Академия Google]
- Уотсон Дж.Д., Крик Ф.Х. (1953). Молекулярная структура нуклеиновых кислот: структура нуклеиновой кислоты дезоксирибозы. Природа 171:737-738. [PubMed] [Академия Google]
- Уильямс А.Л., Джейкобс С.Б., Морено-Масиас Х., Уэрта-Чагоя А., Черчхаус С., Маркес-Луна С., Консорциум по диабету 2 типа SIGMA и др. (2013). Варианты последовательности в SLC16A11 являются распространенным фактором риска развития диабета 2 типа в Мексике. Природа 506:97-101. [Бесплатная статья PMC] [PubMed] [Google Scholar]
 Дети-калеки?
Am J Orthod Oral Surg
28:35-40. [Академия Google]
Дети-калеки?
Am J Orthod Oral Surg
28:35-40. [Академия Google] М., Стоун П.А. (2013). Изучение командного процесса: разработка и поддержание эффективной черепно-лицевой помощи в команде. В: Расщелина губы и неба: диагностика и лечение. Берковиц С., редактор. Берлин, Германия: Springer-Verlag. [Академия Google]
М., Стоун П.А. (2013). Изучение командного процесса: разработка и поддержание эффективной черепно-лицевой помощи в команде. В: Расщелина губы и неба: диагностика и лечение. Берковиц С., редактор. Берлин, Германия: Springer-Verlag. [Академия Google] [PubMed] [Академия Google]
[PubMed] [Академия Google] [PubMed] [Академия Google]
[PubMed] [Академия Google]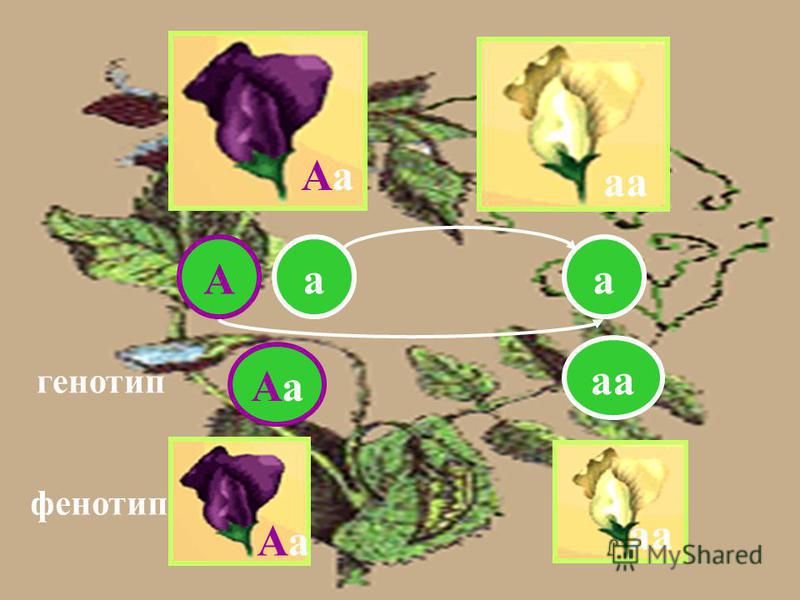 (2014). Будущее исследований в области черепно-лицевой биологии и что это будет означать для профессионального образования и клинической практики в области гигиены полости рта. Ост Дент Дж.
59(1 доп): 1С-5С. [PubMed] [Академия Google]
(2014). Будущее исследований в области черепно-лицевой биологии и что это будет означать для профессионального образования и клинической практики в области гигиены полости рта. Ост Дент Дж.
59(1 доп): 1С-5С. [PubMed] [Академия Google] [PubMed] [Академия Google]
[PubMed] [Академия Google]Статьи из Journal of Dental Research предоставлены Международной и Американской ассоциациями стоматологических исследований
Корреляция генотип-фенотип при болезни Фабри — Болезнь Фабри
Фенотип болезни может модулироваться генетическими и негенетическими модификаторами. Корреляция между генотипом и фенотипом представляет собой статистическую взаимосвязь, предсказывающую физическую особенность человека или аномалию у пациента с данной мутацией или группой подобных мутаций. Анализ корреляций генотип-фенотип при болезни Фабри затруднен рядом факторов, таких как высокая доля частных мутаций, большая фенотипическая гетерогенность, связанная с одной и той же мутацией, как среди больных из одной семьи, так и среди неродственных семей. и тот факт, что у пациентов с болезнью Фабри могут развиться осложнения, связанные с заболеванием, которые наблюдаются с высокой распространенностью в общей популяции. Связанная с генотипом информация о структуре фермента, полученная из кристаллографического анализа, вместе с измерением остаточной активности фермента может помочь в прогнозировании вероятности тяжелого или ослабленного фенотипа. Отдельные генотипы могут иметь фармакогеномное значение; например, максимально возможный клинический ответ на терапию молекулярными шаперонами можно предсказать по корреляции между величиной остаточной активности фермента и ассоциированным клиническим фенотипом. Женские гетерозиготы демонстрируют значительную фенотипическую изменчивость, которую можно было бы лучше понять, учитывая более полные эпидемиологические данные.
Анализ корреляций генотип-фенотип при болезни Фабри затруднен рядом факторов, таких как высокая доля частных мутаций, большая фенотипическая гетерогенность, связанная с одной и той же мутацией, как среди больных из одной семьи, так и среди неродственных семей. и тот факт, что у пациентов с болезнью Фабри могут развиться осложнения, связанные с заболеванием, которые наблюдаются с высокой распространенностью в общей популяции. Связанная с генотипом информация о структуре фермента, полученная из кристаллографического анализа, вместе с измерением остаточной активности фермента может помочь в прогнозировании вероятности тяжелого или ослабленного фенотипа. Отдельные генотипы могут иметь фармакогеномное значение; например, максимально возможный клинический ответ на терапию молекулярными шаперонами можно предсказать по корреляции между величиной остаточной активности фермента и ассоциированным клиническим фенотипом. Женские гетерозиготы демонстрируют значительную фенотипическую изменчивость, которую можно было бы лучше понять, учитывая более полные эпидемиологические данные.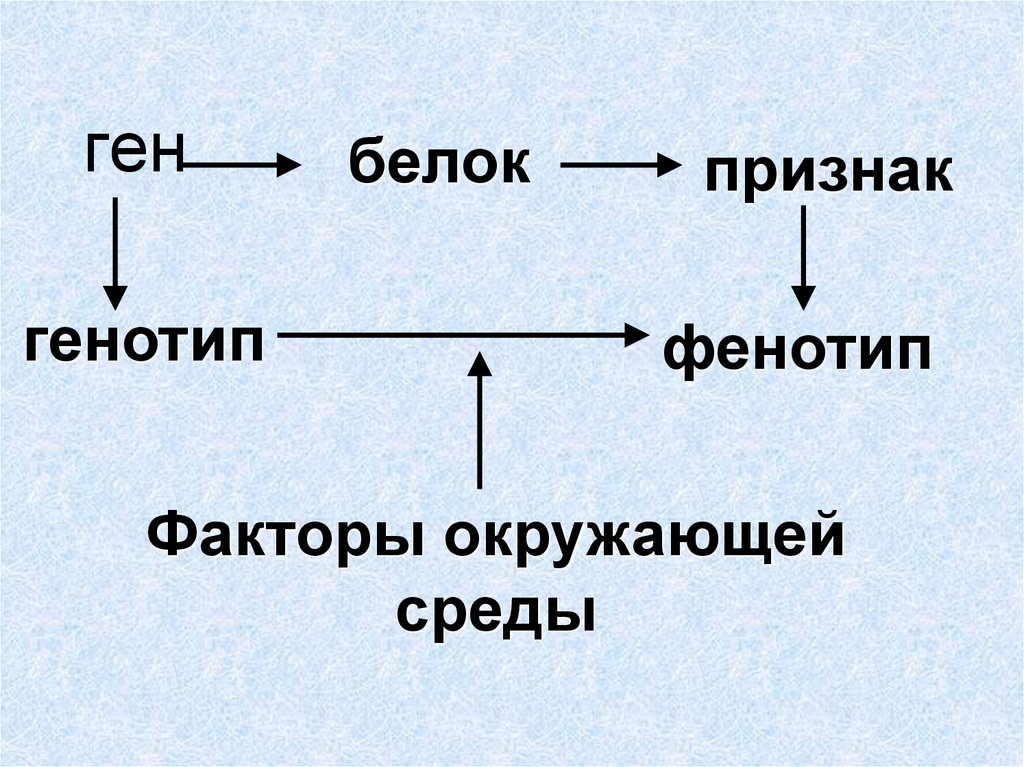
Введение
Корреляция между генотипом (от греческого genos — раса, потомство) и фенотипом (от греческого phaino —, от phainein — показывать) определяется как сверхслучайная вероятность отдельной мутации, связанной с определенной физической особенностью или аномалией. Генотип и фенотип имеют статистическую зависимость. Чем чаще определенный фенотип наблюдается в связи с определенным генотипом, тем выше вероятность того, что неродственный человек с тем же генотипом проявит черты или аномалии, наблюдаемые в популяции, несущей тот же аллель (аллели). Эта взаимосвязь может быть выражена как положительная прогностическая ценность (PPV) данного генотипа для конкретного фенотипа следующим образом:
Болезнь Гоше (OMIM 230800 тип I; OMIM 231000 тип III) представляет собой классический пример установленной корреляции генотип-фенотип при лизосомной болезни накопления. В этом состоянии гомозиготность по аллелю p.Leu483Pro (традиционное обозначение Leu444Pro) имеет высокий PPV для клинически четко определенного фенотипа нейропатического заболевания. С другой стороны, аллель p.Asn409Ser (традиционное обозначение Asn370Ser) имеет высокий PPV для ненейронопатического фенотипа того же расстройства.
С другой стороны, аллель p.Asn409Ser (традиционное обозначение Asn370Ser) имеет высокий PPV для ненейронопатического фенотипа того же расстройства.
Общие и эпидемиологические соображения
Анализу генотип-фенотип при болезни Гоше способствует высокая распространенность в популяции небольшого числа отдельных мутаций, представляющих собой мутации-основатели. Таким образом, фенотип большого числа пациентов можно оценить в когорте, несущей ту же мутацию. Кроме того, фенотип нейропатической болезни Гоше состоит из отчетливых симптомов, редко встречающихся в общей популяции; например, надъядерный паралич взора, атаксия, деменция и миоклоническая эпилепсия.
Анализ корреляций генотип-фенотип при болезни Фабри затруднен по ряду причин. Во-первых, в отличие от болезни Гоше, здесь высока доля частных мутаций; то есть большинство семейств несут разные мутации [1]. Во-вторых, существует высокая степень клинической вариабельности как среди пациентов из одной семьи, так и среди пациентов из неродственных семей с одинаковой мутацией.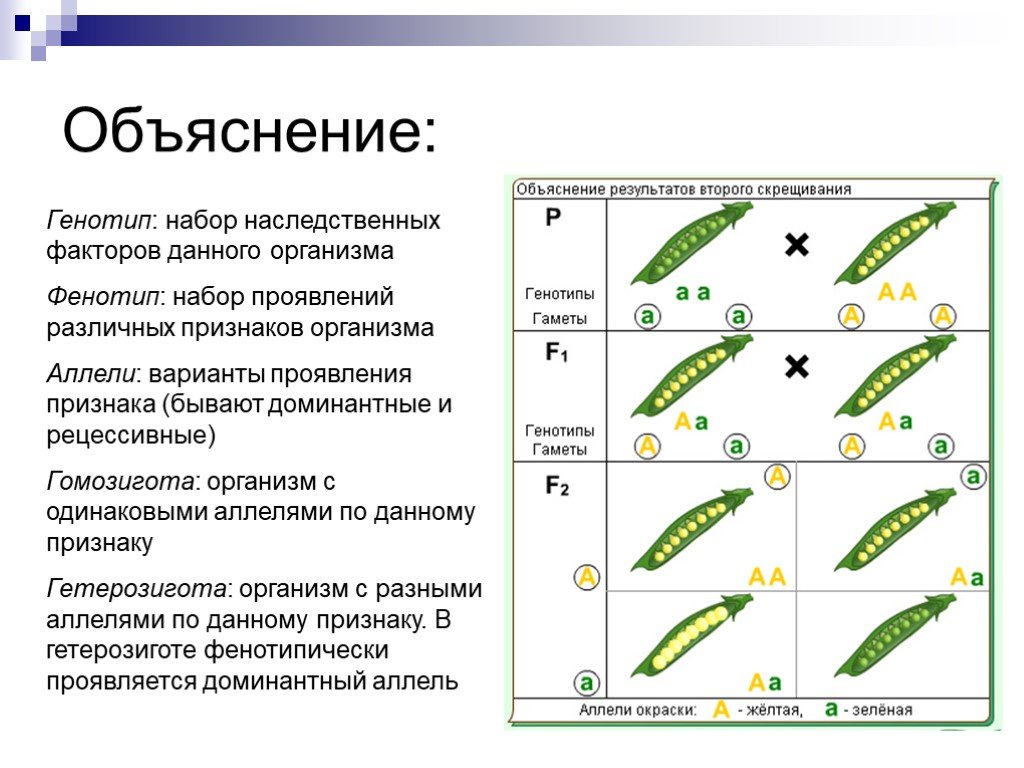 В-третьих, многие клинические признаки болезни Фабри часто наблюдаются у населения в целом, например, невропатическая боль и боль в животе, головная боль, шум в ушах, потеря слуха, диарея и сердечно-сосудистые заболевания. В этом контексте болезнь Фабри можно рассматривать как фактор риска часто встречающейся патологии [2]. В-четвертых, возможно, что часть фенотипа может быть обусловлена негенетическими факторами, такими как медленно прогрессирующее и/или локальное накопление неправильно свернутых мутантных белков, что может привести к образованию нерастворимых агрегатов, что, как предполагалось, объясняет повышенная частота паркинсонизма среди пациентов, гетерозиготных по болезни Гоше [3].
В-третьих, многие клинические признаки болезни Фабри часто наблюдаются у населения в целом, например, невропатическая боль и боль в животе, головная боль, шум в ушах, потеря слуха, диарея и сердечно-сосудистые заболевания. В этом контексте болезнь Фабри можно рассматривать как фактор риска часто встречающейся патологии [2]. В-четвертых, возможно, что часть фенотипа может быть обусловлена негенетическими факторами, такими как медленно прогрессирующее и/или локальное накопление неправильно свернутых мутантных белков, что может привести к образованию нерастворимых агрегатов, что, как предполагалось, объясняет повышенная частота паркинсонизма среди пациентов, гетерозиготных по болезни Гоше [3].
Кристаллографическая модель структуры α-галактозидазы А позволила по-новому взглянуть на корреляции генотип-фенотип при болезни Фабри. Garman и Garboczi [4] нанесли мутаций GLA на трехмерную структурную модель фермента и сопоставили генотип и фенотип с помощью метаанализа фенотипов, описанных в литературе.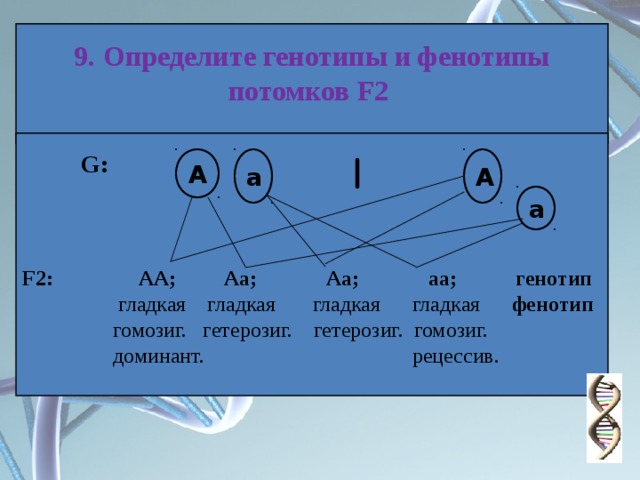 По-видимому, миссенс-мутации, вызывающие мягкий фенотип, затрагивают остатки, которые имеют тенденцию находиться на поверхности белка или легко доступны с поверхности. Напротив, миссенс-мутации, связанные с тяжелой болезнью Фабри, чаще затрагивают остатки, находящиеся вблизи активного центра, или разрушают гидрофобное ядро фермента [4].
По-видимому, миссенс-мутации, вызывающие мягкий фенотип, затрагивают остатки, которые имеют тенденцию находиться на поверхности белка или легко доступны с поверхности. Напротив, миссенс-мутации, связанные с тяжелой болезнью Фабри, чаще затрагивают остатки, находящиеся вблизи активного центра, или разрушают гидрофобное ядро фермента [4].
Остаточная активность фермента и фенотипические варианты
Преждевременный стоп-кодон или мутации, затрагивающие активный сайт фермента, обычно приводят к нефункциональному белку или полному отсутствию генного продукта (функциональный нулевой аллель). Напротив, миссенс-мутация может привести к значительной потере метаболической активности α-галактозидазы А, хотя белок все еще может сохранять некоторую остаточную ферментативную активность [5]. Таким образом, анализ генотип-фенотип при болезни Фабри может быть выполнен путем сопоставления очень изменчивого и индивидуального генотипа с поддающимся количественному определению промежуточным фенотипом, таким как остаточная активность α-галактозидазы А. Для этой цели пациенты могут быть классифицированы как имеющие или не имеющие остаточной ферментативной активности. Для обеспечения качества необходимо, чтобы определение активности α-галактозидазы А in vitro следует проводить в присутствии N -ацетилгалактозамина, ингибитора α-галактозидазы B, чтобы подавить фоновый шум в анализе и избежать ложно завышенных результатов. Пациенты с остаточной активностью фермента были описаны как пациенты с более поздним началом поражения почек, меньшей распространенностью невропатической боли, меньшим количеством дисморфических признаков и более низкой частотой потери слуха по сравнению с лицами без остаточной активности фермента [5–8]. . В естественнонаучном исследовании 25 детей у трех мальчиков с остаточной активностью ферментов были нормальные концентрации глоботриаозилцерамида в крови и моче и не было мутовок роговицы [9].]. Иногда сообщают о пациентах (обычно с остаточной активностью фермента) с сердечными или почечными вариантами болезни Фабри.
Для этой цели пациенты могут быть классифицированы как имеющие или не имеющие остаточной ферментативной активности. Для обеспечения качества необходимо, чтобы определение активности α-галактозидазы А in vitro следует проводить в присутствии N -ацетилгалактозамина, ингибитора α-галактозидазы B, чтобы подавить фоновый шум в анализе и избежать ложно завышенных результатов. Пациенты с остаточной активностью фермента были описаны как пациенты с более поздним началом поражения почек, меньшей распространенностью невропатической боли, меньшим количеством дисморфических признаков и более низкой частотой потери слуха по сравнению с лицами без остаточной активности фермента [5–8]. . В естественнонаучном исследовании 25 детей у трех мальчиков с остаточной активностью ферментов были нормальные концентрации глоботриаозилцерамида в крови и моче и не было мутовок роговицы [9].]. Иногда сообщают о пациентах (обычно с остаточной активностью фермента) с сердечными или почечными вариантами болезни Фабри.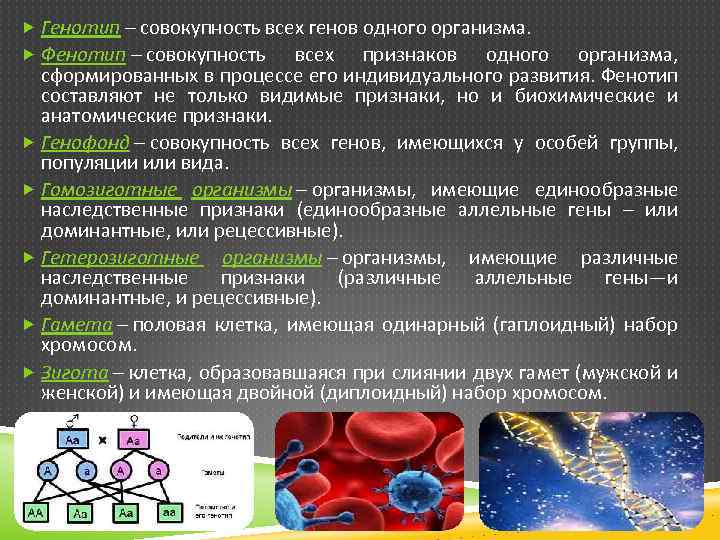 Мы считаем, что эти термины отражают фенотипический «моментальный снимок» в данный момент времени и должны использоваться с осторожностью, поскольку они не принимают во внимание тот факт, что болезнь Фабри является прогрессирующим состоянием. Таким образом, у пациентов с так называемым сердечным вариантом может развиться протеинурия, которая по определению является поражением почечной системы. Кардиальные или почечные варианты, таким образом, отражают широкий спектр клинической гетерогенности болезни Фабри и теоретически могут существовать для любого данного признака или симптома в данном возрасте (4). Таким образом, у пациентов мужского пола оказывается, что наличие остаточной активности сдвигает кривую, представляющую бремя болезни, вправо (). Дальнейшие исследования естествознания людей с остаточной активностью ферментов могут выявить непрерывный спектр фенотипов. Помимо лучшего понимания расстройства, такие исследования потенциально могут помочь в прогнозировании результатов лечения молекулярными шаперонами (см.
Мы считаем, что эти термины отражают фенотипический «моментальный снимок» в данный момент времени и должны использоваться с осторожностью, поскольку они не принимают во внимание тот факт, что болезнь Фабри является прогрессирующим состоянием. Таким образом, у пациентов с так называемым сердечным вариантом может развиться протеинурия, которая по определению является поражением почечной системы. Кардиальные или почечные варианты, таким образом, отражают широкий спектр клинической гетерогенности болезни Фабри и теоретически могут существовать для любого данного признака или симптома в данном возрасте (4). Таким образом, у пациентов мужского пола оказывается, что наличие остаточной активности сдвигает кривую, представляющую бремя болезни, вправо (). Дальнейшие исследования естествознания людей с остаточной активностью ферментов могут выявить непрерывный спектр фенотипов. Помимо лучшего понимания расстройства, такие исследования потенциально могут помочь в прогнозировании результатов лечения молекулярными шаперонами (см. ниже).
ниже).
Рисунок 1
Болезнь Фабри — это фенотипически гетерогенное состояние, при котором поражение основных органов может представлять собой континуум от кажущегося поражения изолированного органа до полиорганных проявлений. На рисунке представлен фенотип с (1) кардиомиопатией, (2) (подробнее…)
Рисунок 2
Достаточный уровень остаточной активности фермента может сместить кривую, отражающую бремя болезни Фабри, вправо. Данные о времени и степени тяжести неполные (обозначены пунктирной линией).
Фармакогеномика
Ферментативная активность у пациентов со специфическими мутациями, такими как p.Arg301Gln и p.Gln279Glu, может быть фармакологически усилена в присутствии аминосахара дезоксигалактоноджиримицина (DGJ) [10]. Препараты этого класса обычно называют молекулярными шаперонами (от французского шаперон , буквально «маленький капюшон», что означает сопровождать). Было показано, что при низких концентрациях эти небольшие молекулы взаимодействуют с ферментом и повышают его активность, тогда как при более высоких концентрациях они обладают ингибирующими свойствами. Чтобы оценить, обладает ли DGJ потенциалом для повышения активности фермента и в какой степени, исследований in vitro необходимы для сравнения активности мутантных ферментов в присутствии и в отсутствие шаперона. Фенотип заболевания пробандов с 10% остаточной активностью фермента, например, может указывать на максимально достижимый уровень улучшения состояния у пациента с низкой остаточной активностью фермента, которая была терапевтически повышена до 10%. При условии, что результаты in vitro трансформируются в клиническую пользу, которая должна быть подтверждена в клинических испытаниях, этот вид терапии может быть полезен для подгрупп пациентов с болезнью Фабри, ассоциированной с определенными генотипами.
Чтобы оценить, обладает ли DGJ потенциалом для повышения активности фермента и в какой степени, исследований in vitro необходимы для сравнения активности мутантных ферментов в присутствии и в отсутствие шаперона. Фенотип заболевания пробандов с 10% остаточной активностью фермента, например, может указывать на максимально достижимый уровень улучшения состояния у пациента с низкой остаточной активностью фермента, которая была терапевтически повышена до 10%. При условии, что результаты in vitro трансформируются в клиническую пользу, которая должна быть подтверждена в клинических испытаниях, этот вид терапии может быть полезен для подгрупп пациентов с болезнью Фабри, ассоциированной с определенными генотипами.
Женские гетерозиготы
При заболеваниях, сцепленных с Х-хромосомой, гетерозиготных женщин обычно называют носителями. Этот термин описывает генетическое состояние гетерозиготности и, следовательно, вероятность передачи данного Х-сцепленного состояния. Однако слово «носитель» по определению не дает никакой информации о фенотипе женского заболевания при Х-сцепленных заболеваниях. Анализируя опубликованные случаи, Dobyns et al. подсчитали, что при болезни Фабри пенетрантность у гетерозиготных женщин составляет 70% (это означает, что 70 из 100 женщин имеют клинические проявления). В том же исследовании клиническая тяжесть заболевания у женщин оценивалась в среднем в 4 балла по шкале от 0 (отсутствие признаков или симптомов) до 100 (полное проявление болезни) [11]. Однако трудно надежно определить пенетрантность и экспрессивность у женщин, гетерозиготных по болезни Фабри, поскольку на момент написания эпидемиологические данные были неполными.
Однако слово «носитель» по определению не дает никакой информации о фенотипе женского заболевания при Х-сцепленных заболеваниях. Анализируя опубликованные случаи, Dobyns et al. подсчитали, что при болезни Фабри пенетрантность у гетерозиготных женщин составляет 70% (это означает, что 70 из 100 женщин имеют клинические проявления). В том же исследовании клиническая тяжесть заболевания у женщин оценивалась в среднем в 4 балла по шкале от 0 (отсутствие признаков или симптомов) до 100 (полное проявление болезни) [11]. Однако трудно надежно определить пенетрантность и экспрессивность у женщин, гетерозиготных по болезни Фабри, поскольку на момент написания эпидемиологические данные были неполными.
Некоторые авторы подняли вопрос о том, является ли болезнь Фабри Х-хромосомным доминантным или рецессивным заболеванием. Согласно принципам, обобщенным Римоином и его коллегами, Х-хромосомное заболевание является доминантным, если фенотип сходен у обоих полов и имеется больше пораженных женщин по сравнению с мужчинами.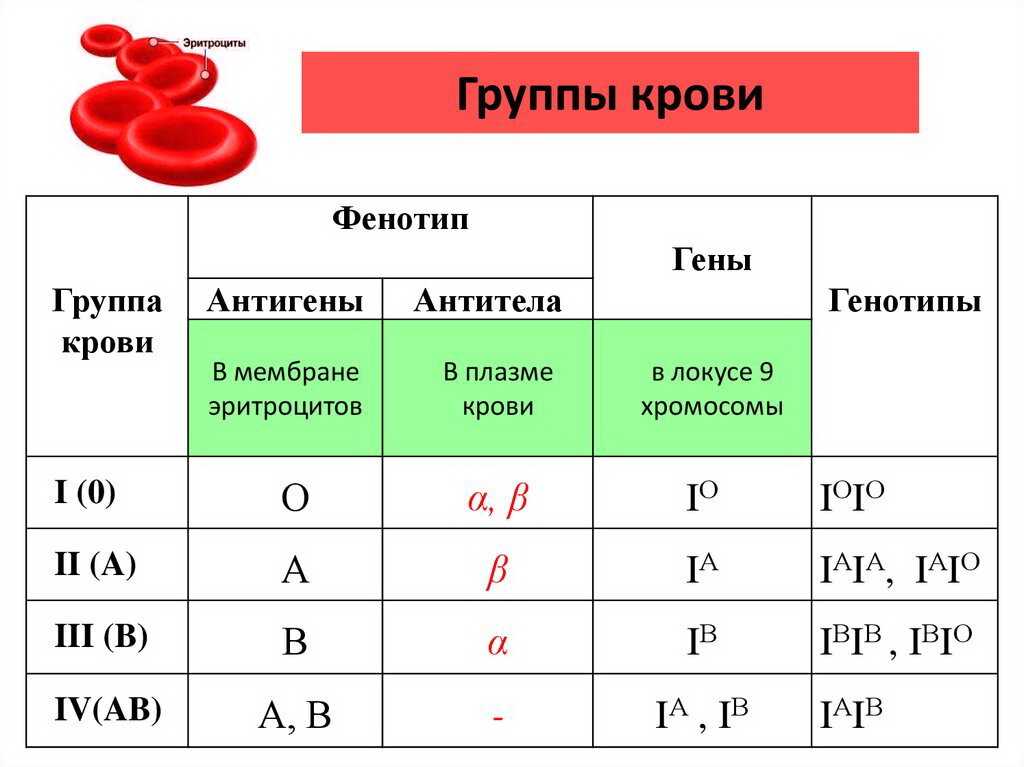 Напротив, при рецессивном признаке поражаются почти исключительно самцы [12]. В регистрах болезней самки и самцы представлены в соотношении примерно 1:1, тогда как по статистике гетерозиготных женщин должно быть в два раза больше, чем гемизиготных мужских. Исследователи, предполагающие, что это состояние является доминантным, будут утверждать, что женщины с болезнью Фабри недостаточно диагностированы, тогда как те, кто предполагает, что это заболевание является рецессивным, будут утверждать, что случаи симптоматических женщин преувеличены. Возможно, что оценка гетерозигот женского пола с легким фенотипом подвержена систематической ошибке; то есть признаки и симптомы не распознаются как проявления болезни Фабри и, следовательно, не относятся к третичным центрам. Обе группы согласны с тем, что, как правило, заболевание начинается позже, а признаки и симптомы, как правило, мягче у женщин, чем у мужчин. Тем не менее, появляется все больше данных, подтверждающих высокую пенетрантность болезни Фабри у женщин-носителей с вариабельным фенотипическим выражением, варьирующим от отсутствия клинических проявлений до классического фенотипа болезни, включая инсульт и почечную недостаточность, как у гемизиготных мужчин [13–19].
Напротив, при рецессивном признаке поражаются почти исключительно самцы [12]. В регистрах болезней самки и самцы представлены в соотношении примерно 1:1, тогда как по статистике гетерозиготных женщин должно быть в два раза больше, чем гемизиготных мужских. Исследователи, предполагающие, что это состояние является доминантным, будут утверждать, что женщины с болезнью Фабри недостаточно диагностированы, тогда как те, кто предполагает, что это заболевание является рецессивным, будут утверждать, что случаи симптоматических женщин преувеличены. Возможно, что оценка гетерозигот женского пола с легким фенотипом подвержена систематической ошибке; то есть признаки и симптомы не распознаются как проявления болезни Фабри и, следовательно, не относятся к третичным центрам. Обе группы согласны с тем, что, как правило, заболевание начинается позже, а признаки и симптомы, как правило, мягче у женщин, чем у мужчин. Тем не менее, появляется все больше данных, подтверждающих высокую пенетрантность болезни Фабри у женщин-носителей с вариабельным фенотипическим выражением, варьирующим от отсутствия клинических проявлений до классического фенотипа болезни, включая инсульт и почечную недостаточность, как у гемизиготных мужчин [13–19]. ].
].
Чтобы более точно отразить распространенность симптоматических Х-сцепленных состояний у женщин, Dobyns et al. предложил использовать только термин «сцепленный с Х-хромосомой» признак, поскольку определения «рецессивный, сцепленный с Х-хромосомой» или «доминантный, сцепленный с Х-хромосомой» не охватывают широкий спектр пенетрантности и экспрессивности [11]. Фенотип гетерозигот может зависеть от комбинации факторов, как в случае их родственников-мужчин, таких как конкретная мутация GLA или аутосомные и/или Х-хромосомные гены-модификаторы (см. ниже). Кроме того, существует ряд проблем, специфичных для гетерозигот, таких как характер лионизации в различных системах органов и варианты последовательности, присутствующие в диком типе 9.0185 Аллель GLA , который также может модифицировать общий фенотип. Для клинического ведения и консультирования гетерозиготных женщин важно признать, что не существует суррогатных маркеров, доступных для прогнозирования риска осложнений, таких как аритмии, инсульт или почечная недостаточность.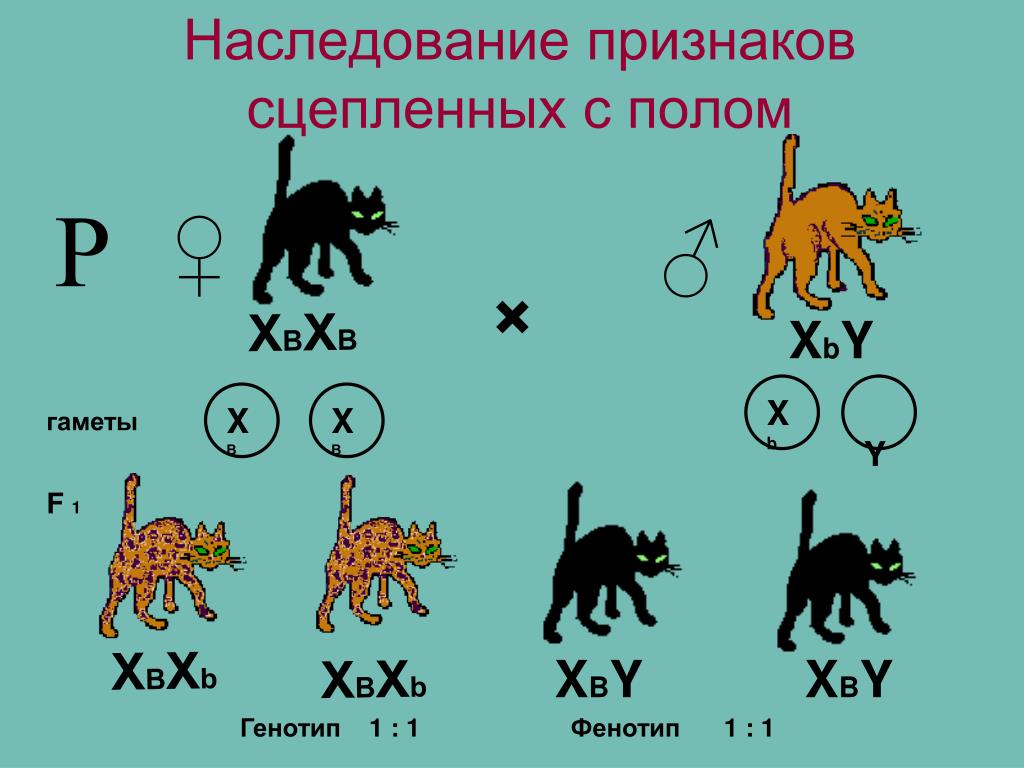
Генетические модификаторы
С точки зрения молекулярной патологии болезнь Фабри можно рассматривать как метаболическую васкулопатию за пределами дефекта одного гена. Следовательно, факторы риска сердечно-сосудистых и цереброваскулярных заболеваний, такие как высокое кровяное давление, повышенный уровень холестерина и злоупотребление никотином, будут неблагоприятно влиять на фенотип у данного пациента. Таким образом, фенотип, вероятно, также модифицируется как генетическими факторами, не связанными с α-галактозидазой А, так и факторами окружающей среды. В недавнем исследовании установлено наличие различных полиморфизмов ДНК генов, кодирующих белки системы воспаления и свертывания крови, такие как интерлейкин 6 (c.−174G>C), p.Glu298Asp эндотелиальной синтазы оксида азота, мутация фактора V p.Arg506Gln и варианты c.-13A>G и IVS6 (интрон F) +79G>A гена ( PROZ ), кодирующего витамин-К-зависимую белка Z, были связаны с повышенным риском поражения головного мозга и инсульта у пациентов с болезнью Фабри [20]. Можно предположить, что относительное влияние генов-модификаторов может быть больше у пациентов с остаточной активностью фермента, чем у пациентов без активности фермента. У пробандов первой группы GLA -неродственные факторы могут по-прежнему оказывать значительное влияние на степень определяемой GLA мутацией остаточной ферментативной активности и, следовательно, на общий фенотип, чего не было бы в случае отсутствия остаточной ферментативной активности.
Можно предположить, что относительное влияние генов-модификаторов может быть больше у пациентов с остаточной активностью фермента, чем у пациентов без активности фермента. У пробандов первой группы GLA -неродственные факторы могут по-прежнему оказывать значительное влияние на степень определяемой GLA мутацией остаточной ферментативной активности и, следовательно, на общий фенотип, чего не было бы в случае отсутствия остаточной ферментативной активности.
Выводы
Многомерные модели, основанные на крупномасштабном систематическом сборе данных, необходимы для получения значимых ассоциаций между генотипом и фенотипом при болезни Фабри. В этом контексте FOS, вероятно, окажется ценным ресурсом, предоставляющим важную информацию для будущих усилий, направленных на определение различных аспектов корреляции генотип-фенотип.
Благодарности
Эта работа была поддержана Программой внутренних исследований Национального института неврологических расстройств и инсульта (Национальные институты здравоохранения, Бетесда, Мэриленд, США).
Ссылки
- 1.
Schäfer E, Baron K, Widmer U, Deegan P, Neumann HP, Sunder-Plassmann G. et al. Тридцать четыре новые мутации гена GLA у 121 пациента с болезнью Фабри. Хум Мутат. 2005; 25:412. [PubMed: 15776423]
- 2.
Schiffmann R, Ries M. Болезнь Фабри – важный фактор риска инсульта. Ланцет. 2005; 366: 1754–6. [PubMed: 16298202]
- 3.
Goker-Alpan O, Schiffmann R, LaMarca ME, Nussbaum RL, McInerney-Leo A, Sidransky E. Паркинсонизм среди носителей болезни Гоше. J Med Genet. 2004;41:937–40. [Бесплатная статья PMC: PMC1735652] [PubMed: 15591280]
- 4.
Garman SC, Garboczi DN. Молекулярный дефект, приводящий к болезни Фабри: структура α-галактозидазы человека. Дж Мол Биол. 2004;337:319–35. [PubMed: 15003450]
- 5.
Брэнтон М.Х., Шиффманн Р., Сабнис С.Г., Мюррей Г.Дж., Квирк Дж.М., Альтареску Г. и др. Естественная история почечной болезни Фабри: влияние активности α-галактозидазы А и генетических мутаций на клиническое течение.
Медицина (Балтимор). 2002; 81: 122–38. [PubMed: 11889412]- 6.
Альтареску Г.М., Гольдфарб Л.Г., Парк К.Ю., Канески С., Джеффрис Н., Литвак С. и др. Идентификация пятнадцати новых мутаций и отношения генотип-фенотип при болезни Фабри. Клин Жене. 2001; 60:46–51. [В паблике: 11531969]
- 7.
Ries M. Количественный анализ невропатических и цереброваскулярных коррелятов потери слуха при болезни Фабри. Школа медицины. Дарем, Северная Каролина: Университет Дьюка; 2005.
- 8.
Райс М., Мур Д.Ф., Робинсон С.Дж., Тиффт С.Дж., Розенбаум К.Н., Брэди Р.О. и другие. Количественная оценка дисморфологии при болезни Фабри. Генет Мед. 2006; 8: 96–101. [PubMed: 16481892]
- 9.
Рис М., Гупта С., Мур Д.Ф., Сачдев В., Квирк Дж.М., Мюррей Г.Дж. и другие. Детская болезнь Фабри. Педиатрия. 2005; 115:e344–55. [В паблике: 15713906]
- 10.
Fan JQ, Ishii S, Asano N, Suzuki Y. Ускоренный транспорт и созревание лизосомальной α-галактозидазы A в лимфобластах Фабри с помощью ингибитора фермента.
Нат Мед. 1999;5:112–5. [PubMed: 9883849]- 11.
Добинс В.Б., Филауро А., Томсон Б.Н., Чан А.С., Хо А.В., Тинг Н.Т. и другие. Наследование большинства признаков, сцепленных с Х-хромосомой, не является доминантным или рецессивным, а только сцепленным с Х-хромосомой. Am J Med Genet A. 2004;129:136–43. [PubMed: 15316978]
- 12.
Римуан Д., Коннор Дж., Пириц Р.Е. Принципы и практика медицинской генетики Эмери и Римуана. Нью-Йорк: Черчилль Ливингстон; 1997, с. 93–94.
- 13.
Guffon N. Клиническая картина у женщин с болезнью Фабри. J Med Genet. 2003;40:e38. [Статья бесплатно PMC: PMC1735431] [PubMed: 12676911]
- 14.
Гупта С., Рис М., Коцопулос С., Шиффманн Р. Связь накопления гликолипидов в сосудах с клиническими проявлениями болезни Фабри: поперечное исследование большой когорты клинически пораженных гетерозиготных женщин. Медицина (Балтимор). 2005; 84: 261–8. [В паблике: 16148726]
- 15.
 Медицина (Балтимор). 2002; 81: 122–38. [PubMed: 11889412]
Медицина (Балтимор). 2002; 81: 122–38. [PubMed: 11889412] Нат Мед. 1999;5:112–5. [PubMed: 9883849]
Нат Мед. 1999;5:112–5. [PubMed: 9883849]